
What is going on with AlphaFold3?
A revolution in the field of Protein Science?
On 8th May 2024, Google Deepmind and Isomorphic Labs introduced the world to their new tool for protein structure prediction, AlphaFold3, a more powerful version of the already existent AlphaFold2, with which Google Deepmind had already reconstructed more than 200 millions protein structures (almost every known protein) and crashed the a priori protein structure prediction challenge that had been chasing Bioinformaticians for decades (I talked about it in more detail here).
Are we on the verge of another revolution? Is AlphaFold3 really a game changer as its predecessor was? In this blog post, we’ll explore the potential breakthroughs and new applications, as well as some limitations that the authors themselves recognized.
What’s new?
If you read the abstract of the paper accepted by Nature and published, open-access, on their website, you will see some interesting news:
The introduction of AlphaFold 2 has spurred a revolution in modelling the structure of proteins and their interactions, enabling a huge range of applications in protein modelling and design. In this paper, we describe our AlphaFold 3 model with a substantially updated diffusion-based architecture, which is capable of joint structure prediction of complexes including proteins, nucleic acids, small molecules, ions, and modified residues. The new AlphaFold model demonstrates significantly improved accuracy over many previous specialised tools: far greater accuracy on protein-ligand interactions than state of the art docking tools, much higher accuracy on protein-nucleic acid interactions than nucleic-acid-specific predictors, and significantly higher antibody-antigen prediction accuracy than AlphaFold-Multimer v2.3. Together these results show that high accuracy modelling across biomolecular space is possible within a single unified deep learning framework.
Let’s break this down, so that Biologists can understand AI concepts and AI Scientists can understand Biology ones:
0. Let’s introduce some terminology
0a. For the Biologists
- Machine Learning: Machine Learning is the process with which computers learn to abstract from some data they have not based on human-made instructions, but on advanced statistical and mathematical models
- Deep Learning: Deep Learning is a Machine Learning framework which is prominently designed on Neural Networks and uses a brain-like architecture to learn.
- Neural Network: A Neural Network is somewhat like a network of neurons in the brain, even though much more simpler: in this sense, there are several checkpoints (the neurons), connected with one another, that receive and pass the information if they reach an activation threshold, exactly as it happens with the action potential of a real neural cell.
0b. For the AI Scientists
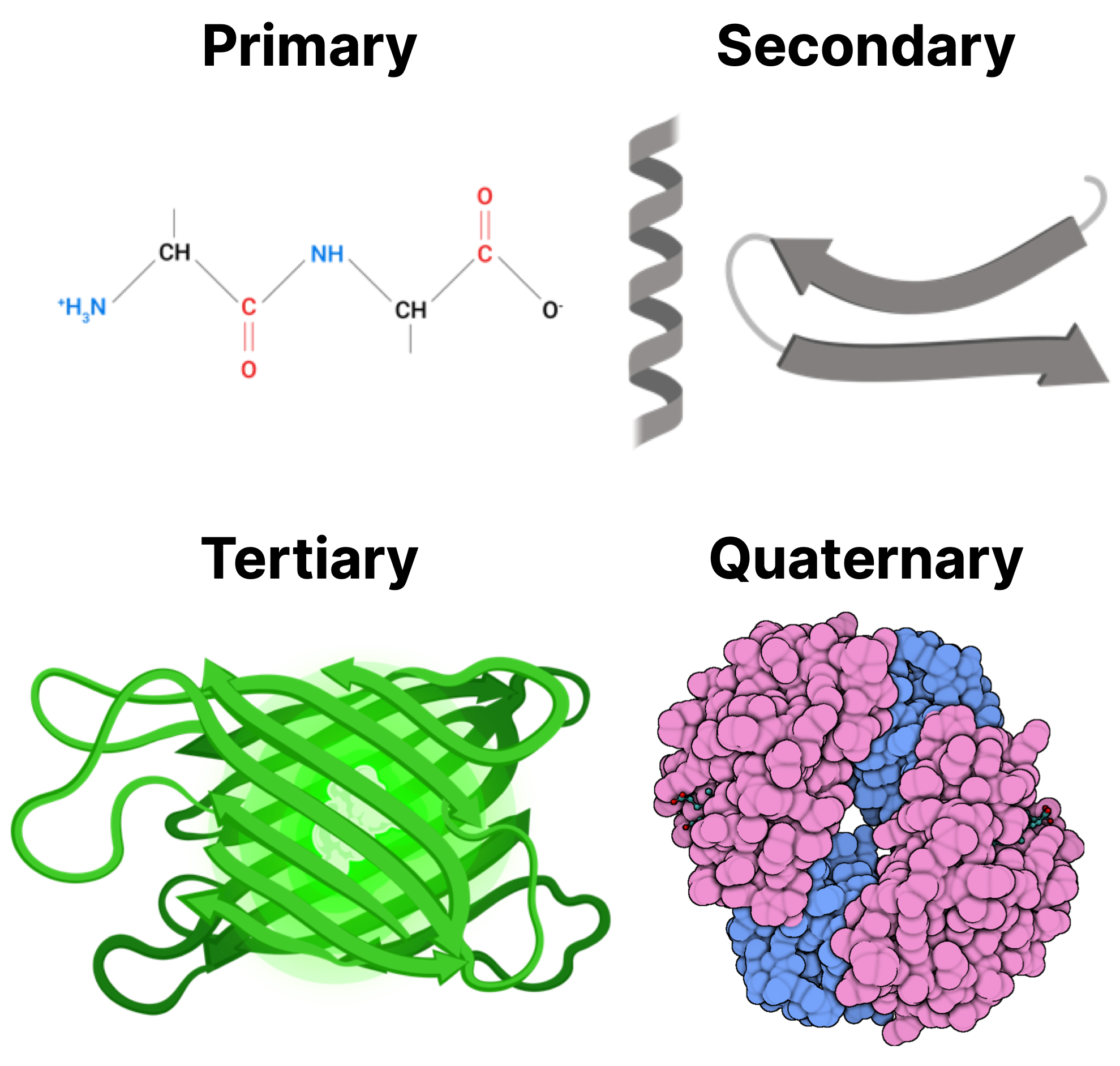
- Protein: Proteins are biomolecules of varying size, made up by little building blocks known as amino acids. They are the factotum of a cell: if you are to imagine a cell as a city, proteins actually represent the transportation system, the communication web, the police, the factory workers… A protein has a primary (flat chain), secondary (mostly 3D but sparse) and tertiary (3D and ordered) structure.
- Ligand: A ligand is something that binds something else: in the context of proteins, it can be a neuro-hormonal signal (like adrenaline) that binds its receptor.
- Nucleic Acids: Nucleic acids (DNA and RNA) are the biomolecules that contain the information about the living system: they are written in a universal language, defined by their building blocks (the nucleotides), and they can be translated into proteins. Thinking of the city example we made before, they could be represented as the Administration Service of it. Nucleic acids often interact with proteins.
1. The diffusion architecture
For diffusion we mean that application of generative AI that is able to create images from a text prompt. The idea behind diffusion is perfectly suitable for the problem of protein structure prediction, as it is a text-based task: indeed, even though the 3D structure of a protein could seem completely unrelated to its 1D amino-acidic chain, there is actually a stronger link than anyone could think of. At the end of the day, indeed, all of the 3D interactions among amino-acids are already defined by their order in the primary chain.
The diffusion architecture in AlphaFold3 receives raw atom coordinates, meaning that, after the first prediction steps coming from a set of neural networks blocks (similar but not equal to those of AlphaFold2), the model is able to turn a “fuzzy” image, with lots of positional and stereochemical noise, to a well-defined and sharp structure. The big advantage of the diffusion model is that it is able to predict the local structure even if the upstream network is not sure about the correct amino-acidic coordinates: this is achieved thanks to the generative process, which is able to produce a distribution of answers that capture most of the possible variability in the protein structure.
As every generative model, also AlphaFold3 diffusion one is prone to hallucination: this is particularly true when it comes to unstructured regions of a protein (that lack a defined and stable tertiary structure), and the AlphaFold3 diffusion blocks are trained in such a way that, in those regions, they produce randomly coiled chains of amino-acids, as done by AlphaFold-Multimeter v2.3 (which generated the images used for hallucination correction training).
2. New tasks and better accuracy
As reported in the abstract, AlphaFold now outperforms task-specific softwares for:
- Protein-Nucleic Acid interaction
- Protein-Ligand interaction
- Antigen-Antibody interaction
Why are these three tasks so important to us?
- Proteins commonly interact with DNA and RNA: as reported by Cozzolino et al. (2021), these interactions “affect fundamental processes such as replication, transcription, and repair in the case of DNA, as well as transport, translation, splicing, and silencing in the case of RNA”. All of these are key cellular functionalities, that, if disrupted, can cause serious diseases. Moreover, understanding how proteins bind DNA and RNA can be really useful in genome editing (CRISPR-Cas9 is actually an RNA-protein-DNA system) and in the fight against bacteria and anti-microbial resistance (lots of the antimicrobial resistance depends on protein-DNA interaction that activates a specific gene which makes the bacterium resistant to the antibiotic).
- Protein-Ligand interaction is key in drug design: up to now, we used “docking”, which means that we simulated the interactions between certain molecule types and proteins by re-iterating those interactions with slightly different chemical structures and positions. Needless to say, this is time-consuming and computationally intense, and AlphaFold3 can definitely improve these aspects, while also retaining a higher accuracy.
- Antigen-Antibody interaction is the process with which some protein produced by our immune system (antibody) bind with foreign or mutated, potentially harmful, molecules: it is one of the methods with which pathogens can be found and eliminated. Predicting these interactions is key in understanding the immune system responses to certain pathogens, but also to something we want to introduce in our body in order to cure it. It also plays an incredibly important role in tumoral cell recognition, as tumoral cell may have some slight modifications of their cell-specific antigen that is not recognized as a threat by our immune system, but can be identified (and thus potentially treated) thanks to computational means.
What are the limitations?
As the authors of the paper reported, they are aware of three big limitations:
- Difficulties in predicting chirality: it is an intrinsic property of a molecule that deals with how the molecule rotates polarized light. Two molecules that differ for nothing but chirality are like your hands: they are perfectly similar, but you can’t superpose them palm to back. Even though some chirality penalty has been introduced, the model still produces about 4% of chirality violating proteins.
- Clashing atoms: there is a tendency, especially with >100 nucleotides nucleic acids interacting with >2000 amino acids proteins, to overlap atoms in the same space region (which is not actually possible).
- Hallucinations, as discussed before, can still happen, so an intrinsic ranking system has been introduced to help the model trashing the hallucinated structures.
- There are still some tasks, such as Antigen-Antibody prediction, where AlphaFold3 can improve. The authors observed improvements when the diffusion models is given more seeds (up to 1000), i.e. a series of numbers that “instruct” the model on how to generate an image, whereas no substantial advancement with more stable diffusion samples.
- As for all protein-prediction models, proteins are predicted in their “static” form, and not “in action”, when they are dynamically inserted into a living cell.
Conclusion and open questions
AlphaFold3 definitely represents a breakthrough in Protein Sciences: still, we are not at an arrival point.
This model marks the kick-off of the new generative AI approach to complex biological problems, which we also saw with OpenCRISPR: on one hand, this holds incredible potential but, on the other, the risk is that we are going to decrease the explainability of our models, leaving scientist with some auto-generated accuracy metrics that are not necessarily able to tell them why a protein has a certain structure.
Another really important topic is that AlphaFold3 is not completely open-source: there is an online-server provided by Google, but the code, as stated in the paper, is not given (except for some mock code that simulates the architecture). This poses a big ethical question: are we sure that we want a world were the access to advanced scientific tools is protected by strict licenses and not everyone can see what is actually going on in softwares by accessing their code?
And, more importantly now than ever, we must ask ourselves: are we really going to rely on non fully open-source AI to design our drugs, deliver targeted genome editing and cure diseases?
References
- Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024). https://doi.org/10.1038/s41586-024-07487-w
- Cozzolino F, Iacobucci I, Monaco V, Monti M. Protein-DNA/RNA Interactions: An Overview of Investigation Methods in the -Omics Era. J Proteome Res. 2021;20(6):3018-3030. doi:10.1021/acs.jproteome.1c00074